
Impacto de las variantes genéticas del SARS-CoV-2 en la hospitalización de pacientes con COVID-19 procedentes de la vigilancia nacional del Ministerio de Salud de Nicaragua, marzo 2020 – septiembre 2022
Impact of SARS-CoV-2 genetic variants on hospitalization of patients with COVID-19 from the national surveillance of the Nicaraguan Ministry of Health, March 2020 - September 2022
Gerald Vásquez Alemán
Instituto de Ciencias Sostenibles, Nicaragua
https://orcid.org/0009-0005-9982-2294
gvasquez@icsnicaragua.org
Christiam Cerpas Cruz
Instituto de Ciencias Sostenibles, Nicaragua
https://orcid.org/0009-0008-9692-5935
ccerpas@icsnicaragua.org
Hanny Moreira Tijerino
Instituto de Ciencias Sostenibles, Nicaragua
https://orcid.org/0009-0008-0701-775X
hmoreira@icsnicaragua.org
Francisco Mayorga-Marín
Universidad Nacional Autónoma de Nicaragua, Managua. Centro de Investigaciones y Estudios de la Salud, UNAN-Managua/CIES, Nicaragua
https://orcid.org/0000-0002-9260-8341
francisco.mayorga@cies.unan.edu.ni
José Juárez
Instituto de Ciencias Sostenibles, Nicaragua
https://orcid.org/0000-0002-0583-478X
jjuarez@icsnicaragua.org
Sonia Arguello
Instituto de Ciencias Sostenibles, Nicaragua
https://orcid.org/0009-0005-0915-5273
sarguello@icsnicaragua.org
Eva Harris
Division of infectious disease and vaccinology, University of California, Berkeley, CA, USA.
https://orcid.org/0000-0002-7238-4037
eharris@berkeley.edu
Aubree Gordon
University of Michigan, Ann Arbor, MI, USA
https://orcid.org/0000-0002-9352-7877
gordonal@umich.edu
Ángel Balmaseda
Instituto de Ciencias Sostenibles, Nicaragua
https://orcid.org/0009-0006-3812-5978
abalmaseda40@gmail.com
RECIBIDO
17/11/2023
ACEPTADO
10/07/2024
RESUMEN
El SARS-CoV-2 se propagó rápidamente por el mundo causando alta morbilidad y mortalidad, por lo que la secuenciación de su genoma se volvió esencial. Numerosos estudios han mostrado cómo las variantes pueden influir en la hospitalización. Este estudio buscó identificar el impacto de las variantes genéticas del SARS-CoV-2 en la probabilidad de hospitalización. Se secuenciaron 1069 muestras de pacientes con COVID-19 en el territorio nacional usando tecnologías de Illumina y Oxford Nanopore. Se realizaron análisis descriptivos y modelos de regresión logística para determinar asociaciones entre las características del virus y el riesgo de hospitalización. Los resultados principales mostraron que las variantes más frecuentes fueron omicrón y Delta (21.32%) y 19B (21.2%). Las menos comunes fueron 21C (Epsilon) y 21H (Mu). Se identificaron mutaciones en las regiones N, M, ORFs y S del genoma del SARS-CoV-2, incluyendo la mutación S:D614G. Se encontraron diferencias significativas en la edad de las personas infectadas con distintas variantes. Hubo una asociación negativa entre el aumento de la edad y el riesgo de infección con la variante 19B, y una asociación positiva con las variantes 20B y Delta. Además, se encontraron asociaciones entre las variantes 20A, 20B, 20C y Delta con el riesgo de hospitalización. La edad fue un factor determinante en el riesgo de infección por ciertas variantes. Algunas variantes aumentaban el riesgo de hospitalización, y la edad, en combinación con las características genéticas de las variantes, influía en este riesgo. El estudio concluye que se identificaron mutaciones de importancia epidemiológica, especialmente la mutación S:D614G, y que ciertas mutaciones y características genéticas del virus pueden conferir mayor riesgo de hospitalización.
PALABRAS CLAVES
SARS CoV-2; COVID-19; secuenciación; RT-PCR
ABSTRACT
SARS-CoV-2 spread rapidly around the world causing high morbidity and mortality, so sequencing its genome became essential. Numerous studies have shown how variants can influence hospitalization. This study sought to identify the impact of SARS-CoV-2 genetic variants on the probability of hospitalization. We sequenced 1069 samples from patients with COVID-19 nationwide using Illumina and Oxford Nanopore technologies. Descriptive analyses and logistic regression models were performed to determine associations between virus characteristics and risk of hospitalization. The main results showed that the most frequent variants were omicron and Delta (21.32%) and 19B (21.2%). The least common were 21C (Epsilon) and 21H (Mu). Mutations were identified in the N, M, ORFs and S regions of the SARS-CoV-2 genome, including the S:D614G mutation. Significant differences were found in the age of infected persons with different variants. There was a negative association between increasing age and risk of infection with the 19B variant, and a positive association with the 20B and Delta variants. In addition, associations were found between the 20A, 20B, 20C and Delta variants with the risk of hospitalization. Age was a determining factor in the risk of infection with certain variants. Some variants increased the risk of hospitalization, and age, in combination with the genetic characteristics of the variants, influenced this risk. The study concludes that epidemiologically important mutations were identified, especially the S:D614G mutation, and that certain mutations and genetic characteristics of the virus may confer increased risk of hospitalization.
KEYWORDS
SARS CoV-2; COVID-19; sequencing; RT-PCR
INTRODUCCIÓN
Desde su identificación por primera vez en diciembre de 2019, el Coronavirus del Síndrome Agudo Respiratorio Severo 2 (SARS-CoV-2) y su enfermedad COVID-19 se ha propagado rápidamente por todo el mundo (Al-Rohaimi & Al Otaibi, 2020), generando 652.2 millones de infecciones y 6.7 millones de muertes globalmente hasta diciembre del 2022 (Organización Panamericana de la Salud, 2022). Dado la importancia del virus y sus consecuencias en hospitalizaciones a nivel global su genoma completo se publicó en febrero del 2020 (Zhu et al., 2020). Como producto de su rápida expansión hacia nuevos países la COVID-19 fue declarada como emergencia sanitaria global por la Organización Mundial de la Salud (OMS) en marzo de 2020 (Organización Mundial de la Salud, 2020).
En plena era genómica y con la aparición de la COVID-19, la secuenciación del SARS-CoV-2 ha demostrado ser una herramienta crucial en la lucha contra la pandemia. El SARS-CoV-2 es un virus que contiene una cadena de ARN en su núcleo, la cual está envuelta por una envoltura proteica. La secuenciación genómica es el proceso de determinar el orden exacto de los nucleótidos en un fragmento de ADN o ARN. En el contexto del SARS-CoV-2, esta tecnología permite obtener y analizar la secuencia del ARN del virus, lo que es esencial para comprender su estructura, y también los cambios que van sucediendo mientras el virus se propaga entre las diferentes poblaciones.
La secuenciación ha permitido obtener las secuencias del genoma del SARS CoV-2 para analizarlo y así identificar y monitorear el surgimiento o introducciones de variantes del virus, facilitando la detección temprana de mutaciones que podrían afectar la eficacia de las vacunas y tratamientos. Además, proporciona datos vitales que ayudan a ajustar las estrategias de respuesta a la pandemia y a diseñar medidas de control más efectivas. La capacidad de secuenciar el genoma del virus real ofrece una ventaja significativa para la vigilancia epidemiológica, permitiendo a los científicos y autoridades sanitarias tomar decisiones informadas y rápidas para mitigar la propagación del virus.
La COVID 19 ha cambiado el escenario epidemiológico mundial y la caracterización genética del SARS-CoV-2 se ha vuelto imprescindible para el manejo adecuado de la enfermedad. Es por ello que, globalmente se han implementado nuevas estrategias en los sistemas de vigilancia genómica mediante la secuenciación del ARN del virus (Brito et al., 2022), lo cual hace que su vigilancia genómica sea imprescindible para las investigaciones epidemiológicas, con el fin de comprender el impacto de los cambios genéticos en los pacientes con COVID-19.
Numerosos estudios han demostrado cómo las variantes genéticas pueden influir en el estado de salud de las personas infectadas y en la evolución de la pandemia en diferentes regiones (Butt et al., 2022; Maslo et al., 2022; Nonaka et al., 2021). Se ha logrado determinar cómo la introducción de nuevas variantes genéticas en la población provoca el surgimiento de nuevas olas de contagio (El-Shabasy et al., 2022).
También, se ha observado que las personas infectadas con la variante ómicron tienen una disminución significativa en el riesgo de desarrollar enfermedad severa en comparación con las personas infectadas por la variante Delta (Van Goethem et al., 2022). Además, se ha observado que las introducciones de nuevas variantes pueden asociarse con cambios en el riesgo de infección y severidad en diferentes grupos de edades (Freitas et al., 2021).
Comprender los factores genéticos asociados a la hospitalización ha podido proporcionar información crucial sobre la patogénesis del COVID-19 y ha sido útil para orientar los esfuerzos que permiten identificar a las personas con mayor riesgo de enfermedad grave, a medida que las nuevas variantes emergen y se propagan en la población. En Nicaragua desde julio de 2021 se ha podido establecer el sistema de secuenciación genómica para caracterizar las variantes circulantes del SARS-CoV-2. Estos datos han proporcionado información clave para mejorar nuestro entendimiento de la respuesta inmunológica en niños con COVID-19 (Maier et al., 2022).
Al investigar los factores genéticos asociados a la hospitalización, se espera contribuir a un mejor entendimiento sobre la COVID-19 y sobre la epidemiología molecular de las variantes del SARS-CoV-2 en Nicaragua, además proporcionar información valiosa para futuras estrategias de investigación, prevención y control. Comprender cómo las variantes genéticas del virus influyen en la gravedad de la enfermedad y en la necesidad de hospitalización puede ayudar a identificar a las poblaciones más vulnerables y a desarrollar medidas más eficaces para protegerlas.
El presente estudio tiene como objetivo analizar las características genéticas del SARS-CoV-2 en toda la región de Nicaragua y determinar cómo estas características están asociadas con la probabilidad de hospitalización.
MATERIALES Y MÉTODOS
Se realizó un estudio de enfoque cuantitativo debido a que permite analizar datos numéricos y establecer relaciones estadísticas entre las variables estudiadas. El estudio fue observacional analítico, ya que se centró en observar y analizar características existentes sin intervenir en el ambiente o en los sujetos del estudio.
Este enfoque permitió analizar las características genéticas de SARS-CoV-2 en toda la región de Nicaragua e identificar características inherentes del virus y la probabilidad de estas con la hospitalización empleando secuencias de nucleótidos del SARS CoV-2 obtenidas mediante secuenciación genómica a partir de muestras de pacientes con COVID 19. El período de estudio es de 2 años y 6 meses y comprende desde la identificación de los primeros casos en el país en marzo del 2020 hasta septiembre del 2022.
La unidad de análisis fue compuesta por secuencias de nucleótidos obtenidas mediante secuenciación genómica de muestras respiratorias obtenidas de Centros de atención primaria y Hospitalaria del sistema público de la red del Ministerio de Salud a nivel nacional y que son parte del sistema de vigilancia nacional de COVID-19. El universo del estudio es igual a la muestra y estuvo compuesto por 1069 secuencias de nucleótidos. La muestra incluyó secuencias de nucleótidos del SARS-CoV-2 obtenidas mediante secuenciación genómica, de distintas áreas geográficas y demográficas del país.
En el estudio se incluyeron las secuencias nucleotídicas del genoma completo obtenidas de pacientes confirmados con COVID-19 y que pasaron filtros correspondientes de control de calidad. Se excluyeron las secuencias con baja cobertura y calidad. Además, se excluyeron las secuencias que provenían de muestras que no cumplen con los estándares de calidad establecidos para una adecuada secuenciación genómica.
Las secuencias genómicas del SARS-CoV-2 se obtuvieron utilizando tecnologías de secuenciación de alto rendimiento, específicamente Illumina y Oxford Nanopore. Una vez completada la secuenciación, la información de los resultados fue registrada en hojas de trabajo. Estas hojas, junto con los resultados del análisis bioinformático, fueron almacenadas en bases de datos específicas. Para el estudio, se utilizó un enfoque de análisis de datos secundarios, empleando estas bases de datos que también incluían información relevante de las muestras. Ninguno de los datos empleados, contenía información que permitiera identificar a los pacientes ya que todos los datos eran secundarios y provenían de la vigilancia genómica del país.
Etapas de la investigación
Etapa 1: Vigilancia epidemiológica
Las muestras secuenciadas son procedentes de la vigilancia nacional de SARS-CoV-2 del Ministerio de Salud (MINSA). Las muestras son tomadas en centros asistenciales de primer, segundo o tercer nivel en todo el territorio Nacional junto una ficha epidemiológica con datos de la muestra y del estado del paciente. Estas muestras fueron transportadas desde cada centro asistencial hacia los Sistema Local de Atención Integral en Salud (SILAIS) y desde aquí, con previa autorización, fueron transportadas en cadena de frío hasta el Laboratorio Nacional de Virología en el Centro Nacional de Diagnóstico y Referencia (CNDR) ubicado en el Complejo Nacional de Salud Concepción Palacios en la capital de Nicaragua, Managua.
Figura 1
Flujo del muestreo de la vigilancia epidemiológica Nacional de COVID-19
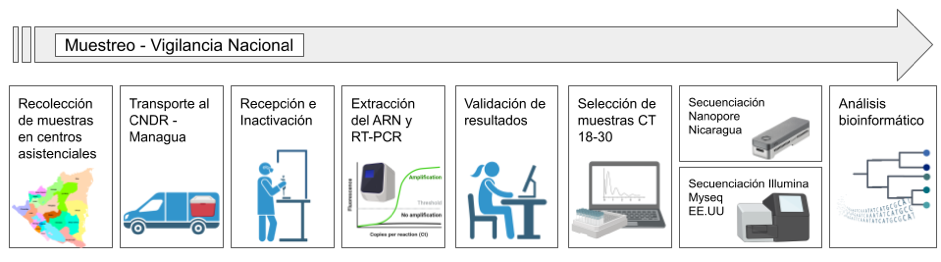
Fuente: Realización propia
Etapa 2: Diagnóstico molecular
En el CNDR, las muestras fueron recepcionadas e inactivadas, posteriormente se realizó un RT-PCR en tiempo real bajo el protocolo de PCR Multiplex estandarizado por el instituto de virología del Hospital Universitario de Charité (Corman et al., 2020), bajo la plataforma ABI 7500 Fast PCR platform (Applied Biosystems, Foster, CA, EE. UU) para su posterior validación. Durante todo el proceso preanalítico y analítico, las muestras y ARN viral fueron manipulados en cabinas de bioseguridad tipo II y con todos los equipos de protección personal requeridos.
Etapa 3: Secuenciación
Para identificar las variantes del SARS-CoV-2 y las mutaciones, se secuenciaron 1069 muestras recolectadas entre marzo de 2020 y septiembre de 2022 (505 muestras secuenciadas en el Laboratorio Nacional de Virología de Nicaragua y 564 en el Hospital Escuela Mount Sinai en Nueva York). En el Laboratorio Nacional de Virología, en Nicaragua, se seleccionaron muestras con un valor de Ct (threshold cycle) entre 18 y 30 del RT-PCR de diagnóstico. El valor de Ct se refiere al número de ciclos necesarios para que la fluorescencia generada en la PCR cruce un umbral específico, indicando la cantidad de material genético viral presente en la muestra.
Posteriormente, se realizó una extracción de ARN utilizando el kit QIAGEN QIAamp® Viral RNA Mini. Para llevar a cabo la secuenciación se utilizó el protocolo de secuenciación de la red Artic (Artic Network, 2020) desde julio del 2020 hasta julio del 2021. A partir de agosto del 2022 se cambió el protocolo de trabajo de la red ARTIC por el protocolo Midnight de Oxford nanopore Technology (Oxford Nanopore Technolgies, 2021) (Figura 2).
Figura 2
Esquema de trabajo para secuenciación usando los protocolos de la red Artic y Midnigth que utiliza el Ministerio de Salud en Nicaragua.
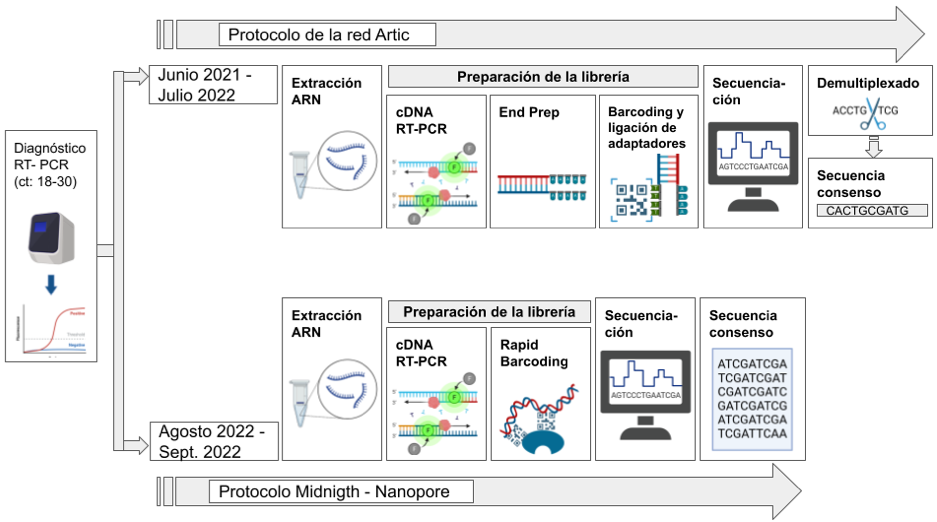
Fuente: Realización propia
Secuenciación usando el protocolo de la red Artic: El ARN fue transcrito inversamente y amplificado utilizando un método de PCR multiplexado a partir de 96 pares de cebadores dirigidos a amplificar todo el genoma del SARS-CoV-2, este proceso generó amplicones de aproximadamente 400 pares de bases. Los amplicones se prepararon para la secuenciación nanopore con el kit de ligación SQK-LSK109 (Oxford Nanopore Technologies, Oxford, Reino Unido) y el kit ONT Native Barcoding Expansion (EXP-NBD196) para etiquetar cada muestra con un marcador molecular. Las librerías fueron cargadas en flowcells FLO-MIN106 R9.4.1 y secuenciadas en un dispositivo MinION Mk1B.
Se utilizó el software RAMPART (v1.0.6) para monitorear el rendimiento de la secuenciación en tiempo real en todos los amplicones. La secuenciación se llevó a cabo en un entorno Ubuntu 18.04 con el software Minknow.
Secuenciación usando el protocolo Midnigth: Este protocolo utiliza un set de cebadores que generan amplicones de entre 900 - 1200 pares de bases. Estos amplicones se utilizaron para preparar una librería con el kit de etiquetado rápido SQK-RBK110.96. El procedimiento de secuenciación fue el mismo que el utilizado con la red ARTIC.
Generación de una secuencia consenso: Durante el periodo en el que se utilizó el protocolo de la red ARTIC, una vez acabó la secuenciación se generaron archivos en formato fastq que contenía los datos crudos de la secuenciación. Se utilizó el software porechop (Wick, 2017/2022) para demultiplexar las lecturas generadas y eliminar los segmentos que contenían información de los cebadores utilizados durante el PCR. Posteriormente se utilizó el software Medaka (Nanoporetech, 2017/2022) para generar secuencias consensos.
Cuando se utilizó el protocolo Midnigth, para para generar secuencias consenso se hizo uso del workflow artic (ARTIC SARS-CoV-2 Workflow, 2021/2023) incluido en la plataforma epi2me y desarrollado por ONT, este software eliminó la información de los cebadores y generó las secuencias consenso. En ambos casos las lecturas se alinearon al genoma de referencia Wuhan-Hu-1 (MN908947.3).
Etapa 4: Análisis de datos
Para analizar las secuencias se utilizó la plataforma Nexstrain, la cual analizó los cambios en los genomas secuenciados clasificando el linaje al que pertenecen. La plataforma clasifica los linajes en clados, pero también brinda la clasificación de la variante con respecto a la clasificación de la Organización Mundial de la salud, la cual clasifica las variantes con las letras del alfabeto griego. En la presente investigación se le nombra “variantes” a todos aquellos linajes que poseen una clasificación por la OMS, y se las denomina “pre-variantes” a aquellas que no poseen una clasificación por la OMS, pero que tienen un clado asignado por la plataforma Nexstrain. Además, la plataforma Nexstrain brinda las mutaciones presentes en cada uno de los genomas analizados.
La información de los linajes del SARS-CoV-2 e información demográfica fue analizada con el lenguaje R (Ihaka & Gentleman, 1996) y el entorno de desarrollo integrado RStudio. Se realizaron tablas de frecuencia para mostrar las características de la muestra. Se construyeron modelos de regresión logística univariada y multivariada para estimar asociaciones (Thulin, 2021).
Las odds ratios (OR) se calcularon exponenciando los coeficientes de regresión obtenidos del modelo de regresión logística. Esto se hace porque los coeficientes representan el logaritmo natural de los odds ratios; por tanto, la exponenciación transforma estos valores logarítmicos a la escala original de odds, proporcionando una interpretación más intuitiva de los efectos del modelo en términos de la probabilidad relativa de los resultados.
Con los resultados de las regresiones se construyeron gráficos de forest plots, donde la línea horizontal paralela al eje y, (en el valor 1 del eje x) representa el valor de referencia o punto neutro. Los puntos en el gráfico representan los OR para cada variante, y los segmentos que se extienden desde cada punto indican los intervalos de confianza correspondientes. Los intervalos de confianza ayudan a evaluar la precisión de la estimación y proporcionan una medida de la incertidumbre asociada al OR. Los segmentos marcados con asteriscos tuvieron un valor de p menor a 0.05 (*p<0.05, **p<0.01, ***p<0.001). Todos los modelos se construyeron bajo un nivel de confianza del 95%.
Limitaciones del estudio
Existe una influencia potencial del sesgo de muestreo de la vigilancia nacional en la investigación. La manera que se realizaba el muestreo por parte del Ministerio de Salud pudo cambiar debido a los cambios de la situación epidemiológica del país, lo que puede introducir sesgos en la representación de la muestra en la presente investigación.
RESULTADOS Y DISCUSIÓN
La secuenciación genómica permitió analizar el genoma completo del SARS CoV 2 en Nicaragua y relacionar el resultado del análisis genómico con características del paciente. La tabla 1 muestra la distribución de las diferentes pre-variantes y variantes del SARS-CoV-2 identificadas mediante secuenciación genómica en relación con el sexo y la hospitalización. Esta investigación identificó un total de 1069 variantes del virus SARS-CoV-2. La variante más frecuente fue Omicron, representando el 27.97% del total. Le siguieron las variantes Delta (21.32%) y 19B (21.2%).
Las variantes menos comunes fueron 21C (Epsilon) y 21H (Mu), con una frecuencia del 0.37% cada una. En cuanto al sexo de los pacientes, se observó que el 41.62% eran mujeres y el 57.99% eran hombres. Hubo un pequeño porcentaje (0.37%) en el que no se registró el sexo. El 31.4% de las muestras secuenciadas provienen de pacientes hospitalizados mientras que un 37.32% de las muestras provienen de pacientes no hospitalizados. No se logró obtener datos de hospitalización en 31.24% del total de las muestras analizadas.
La distribución de las variantes identificadas en este estudio parece ser heterogéneas con respecto al número. Variantes como Omicron, Delta o la pre-variante del clado 19B sobresalen en número con respecto a las demás. Esto puede ser atribuible al período en el que circularon las variantes y pre-variantes. Diversos autores muestran que para el 2020 hubo una especie de competencia entre las variantes y pre-variantes circulantes y el número total de casos era provocado por fracciones de distintos linajes en las que se encuentran las pre-variantes 19A, 19B, 20A, 20B y 20C durante el 2020 y principios del 2021 en Países como Inglaterra, pero también países de la región como Costa Rica y Colombia (Castañeda et al., 2021; Mishra et al., 2021; Molina-Mora et al., 2021).
En la presente investigación, Delta y Omicrón son las variantes que reportan mayor número de observaciones, al igual que los datos reportados por la plataforma GISAID hasta el 2022 a nivel mundial donde estas variantes son las responsables de los brotes epidémicos más importantes (GISAID, 2022).
Tabla 1
Distribución de variantes identificadas en 1069 genomas del SARS-CoV-2 por sexo y estado de hospitalización de los pacientes.
| Clado (Variante) | n (%) |
Sexo n (%) |
Hospitalización n (%) |
||||
|---|---|---|---|---|---|---|---|
| Masculino | Femenino | Sin dato | Si | No | sin dato | ||
| 19A | 1 (0.09%) |
0 | 1 (0.09%) | 0 | 0 | 0 | 1 (0.09%) |
| 19B | 227 (21.2%) |
97 (9.07%) | 130 (12.16%) | 0 | 69 (6.45%) | 141 (13.18%) | 17 (1.59%) |
| 20A | 16 (1.49%) |
8 (0.74%) | 8 (0.74%) | 0 | 11 (1.02%) | 5 (0.46%) | 0 |
| 20B (Zeta) | 119 (11.13%) |
52 (4.86%) | 67 (6.26%) | 0 | 59 (5.51%) | 54 (5.05%) | 6 (0.56%) |
| 20C | 39 (3.64%) |
20 (1.87%) | 19 (1.77 %) | 0 | 25 (2.33%) | 14 (1.31%) | 0 |
| 20H (Beta, V2) | 1 (0.09%) |
0 | 1 (0.09%) | 0 | 0 | 1 (0.09%) | 0 |
| 20I (Alpha, V1) | 3 (0.28%) |
2 (0.18%) | 1 (0.09%) | 0 | 2 (0.18%) | 1 (0.09%) | 0 |
| 20J (Gamma, V3) | 88 (8.23%) |
39 (3.64%) | 48 (4.49%) | 1(0.09%) | 24 (2.24%) | 32 (2.99%) | 32 (2.99%) |
| 21C (Epsilon) | 4 (0.37%) |
3 (0.28%) | 1 (0.069%) | 0 | 1 (0.09%) | 3 (0.28%) | 0 |
| 21G (Lambda) | 7 (0.64%) |
5 (0.46%) | 2 (0.18%) | 0 | 0 | 3 (0.28%) | 4 (0.37%) |
| 21H (Mu) | 4 (0.37%) |
1 (0.09%) | 3 (0.28%) | 0 | 0 | 4 (0.37%) | 0 |
| Delta | 228 (21.32%) |
100 (9.3%) | 128 (11.97%) | 0 | 88 (8.29%) | 58 (5.42%) | 82 (7.67%) |
| Omicron | 299 (27.97%) |
111 (10.38%) | 185 (17.3%) | 3 (0.28%) | 42 (3.92%) | 67 (6.26%) | 190 (17.7%) |
| recombinant | 33 (3.08%) |
7 (0.65%) | 26 (2.43%) | 0 | 15 (1.4%) | 16 (1.49%) | 2 (0.18%) |
| Totales | 1069 (100%) |
445 (41.62%) | 620 (57.99%) | 4 (0.37%) | 336 (31.4%) | 399 (37.32%) | 334 (31.24%) |
Fuente: Base de datos de resultados secuenciación del Laboratorio Nacional de Virología, Ministerio de Salud
La media de edad por cada grupo de pacientes infectados por diferentes variantes rondó entre los 37 y 55 años (Figura 3). La distribución de las edades se comportó de manera similar en la mayoría de los grupos. La frecuencia es más alta en el grupo de adultos jóvenes (20 a 40 años aproximadamente).
Se realizó el test de Shapiro Willk para determinar la normalidad de la distribución de la edad, donde se determinó que la edad en los grupos donde la n es más grande hay una distribución normal (Ver significancias en la figura 3), pero las muestras más pequeñas tienden a manifestar una distribución no normal. Al realizar el test de Kruskal Wallis para determinar diferencia de medias entre los diferentes grupos se encontró que hay diferencias entre las edades de los pacientes infectados con las distintas variantes (p =0.005).
Los resultados de nuestro análisis revelan una variabilidad en la edad de los pacientes infectados por diferentes variantes del virus, con medias de edad que oscilan entre los 37 y 55 años, lo cual podría estar relacionado con factores de exposición y comportamiento social característicos de este grupo etario. La prueba de Shapiro-Wilk mostró que, en los grupos con tamaños de muestra más grandes, la distribución de la edad sigue una distribución normal, mientras que, en grupos más pequeños, la distribución tiende a no ser normal.
Esta heterogeneidad sugiere que factores socio-demográficos y biológicos específicos de cada grupo pueden influir en la distribución de la edad. El resultado significativo del test de Kruskal-Wallis (p = 0.005) indica que existen diferencias estadísticamente significativas en la edad de los pacientes infectados con distintas variantes del virus. Desde una perspectiva epidemiológica, este hallazgo es crucial, ya que sugiere que ciertas variantes podrían estar afectando de manera desproporcionada a diferentes grupos de edad, posiblemente debido a diferencias en la transmisibilidad, patogenicidad, o respuestas inmunológicas entre grupos etarios.
Figura 3
Distribución de la edad de los pacientes según las variantes identificadas mediante secuenciación genómica.
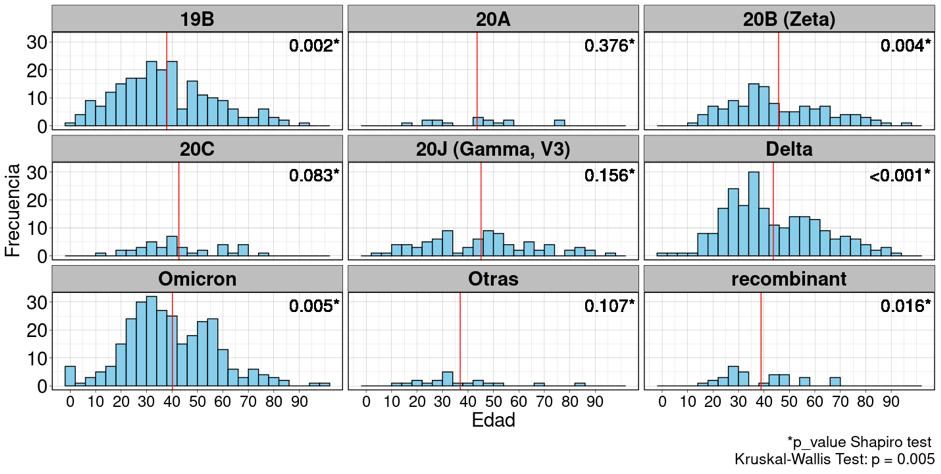
Fuente: Base de datos de resultados secuenciación del Laboratorio Nacional de Virología, Ministerio de Salud
La secuenciación permitió identificar y cuantificar las 30 mutaciones más frecuentes en el conjunto de genomas de las variantes (Figura 4). Entre estas 30 mutaciones se identificaron mutaciones en distintas las regiones del genoma del SARS-CoV-2, como en la región M (que codifica para la proteína de membrana), la región N (que codifica para la proteína de la nucleocápside), los genes ORF1a, ORF1b y ORF3a (marcos de lectura abierto) y genes que codifican para la proteína S (Spike).
Solamente se identificó una mutación en la región M, 8 mutaciones en la región N, se identificaron 5 mutaciones en la región S y en las regiones de los genes ORF se identificaron la mayoría de las mutaciones, siendo la región ORF1a la región ORF con más mutaciones (n= 8), con la misma cantidad de mutaciones de la región N.
En este estudio, de las mutaciones identificadas, sobresalen en frecuencia las mutaciones ORF1a:T3255I identificada en 372 genomas, en los genes ORF1b las mutaciones P1000l y P314L identificadas en 334 y 402 genomas respectivamente. En la región S las mutaciones más sobresalientes en frecuencia son L452R con una frecuencia de 357 y la mutación D614G encontrada 628 genomas (58.7% presente en los genomas). Esta última destaca por ser la mutación que se observó con más frecuencia en todos los genomas analizados.
Un meta-análisis en el que se extrajo información de bases de datos que contenían datos de 165 estudios de mutaciones del SARS-CoV-2 encontró mutaciones en todas las regiones del genoma, al igual que en este estudio (Farhud & Mojahed, 2022). El análisis de las mutaciones identificadas en el genoma del SARS-CoV-2 revela también una amplia diversidad genética en la población de variantes identificadas. Las mutaciones más frecuentes se distribuyen en diferentes regiones del genoma, incluyendo los genes M, N, ORF1a, ORF1b, ORF3a, ORF8 y S. Una de las mutaciones más sobresalientes es la mutación S:D614G que está presente en todas las variantes y pre-variantes. Varios investigadores han encontrado asociaciones entre esta mutación y la hospitalización, además de un aumento en la tasa de infectividad (Korber et al., 2020). Sin embargo, es curioso que esta mutación está presente en la mayoría de los linajes, incluso aquellas que no muestran asociación con la hospitalización.
También se identificó una frecuencia predominante de mutaciones en los genes de marco de lectura abierto (ORF). Previamente se ha documentado que las mutaciones en estos genes pueden modificar el comportamiento del SARS-CoV-2 pues están involucrados en el ensamblaje de proteínas no accesorias que pueden aportar a que el virus pueda evadir la respuesta inmune y aumentar su tasa de infectividad (Prüβ, 2022). Todos estos factores genéticos pueden tener un impacto en la severidad y hospitalización.
Figura 4
Top 30 de las mutaciones identificadas más frecuentemente mediante secuenciación genómica en las variantes del SARS-CoV-2 en 735 pacientes con dato de hospitalización
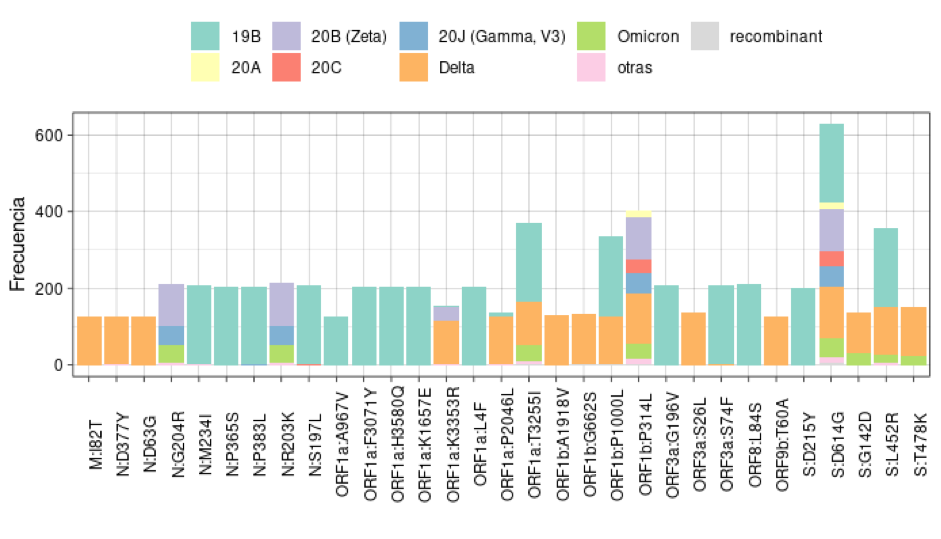
Fuente: Base de datos de resultados secuenciación del Laboratorio Nacional de Virología, Ministerio de Salud
Para ir más allá de los análisis meramente descriptivos, se usó un enfoque de regresión logística univariada para evaluar los datos de edad y variante con la finalidad de determinar si la edad está relacionada estadísticamente con la probabilidad de infección con alguna variante (figura 5). La pre-variante del clado 19B mostró una asociación estadísticamente significativa con sentido negativo con la edad, con un Odd Ratio (OR) de 0.93 (Intervalo de Confianza del 95%: 0.89 - 0.935). Esto indica que a medida que la edad aumenta en 5 años la probabilidad de ser infectado con la pre-variante del clado 19B disminuye, este hallazgo fue altamente significativo, con un valor p menor que 0.001.
Por otro lado, la pre-variante del clado 20B y la variante Delta mostraron asociaciones significativas con la edad. 20B posee un OR de 1.07 (Intervalo de Confianza del 95%: 1.01 - 1.12), lo que indica un aumento en el chance de ser infectado por esta pre-variante cuando la edad aumenta en 5 años. La variante Delta también mostró significancia estadística en el aumento de la edad, con un OR de 1.04 (Intervalo de Confianza del 95%: 1.02 - 1.08). Estos hallazgos fueron respaldados por valores de p significativos de 0.008 y 0.04, respectivamente.
Las pre-variantes 20A, 20C, y la variante Omicrón y además de las variantes recombinantes no mostraron asociaciones significativas con la edad. Sus valores de p no fueron significativos, lo que sugiere que no hay diferencias sustanciales en la probabilidad de ciertas edades entre las personas infectadas con estas variantes.
Este análisis muestra que la probabilidad de ser infectado con la pre-variante del clado 19B disminuye mientras la edad aumenta. La pre-variante 19B acumula una diversidad amplia de mutaciones. Esto es un estilo de huella molecular que indica que esta pre-variante circuló por un tiempo prolongado en la población y esto provocó el desarrollo de un alto número de mutaciones en su genoma. Esta exposición prolongada pudo influir en el desarrollo de la inmunidad en la población expuesta y una consecuente protección, a como sugieren nuestros resultados.
Figura 5
Regresión logística univariada que muestra las asociaciones entre las variantes y la edad (en períodos de 5 años)
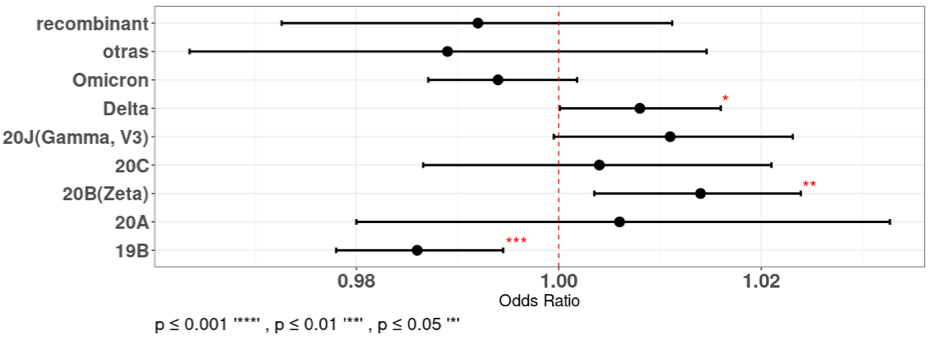
Fuente: Base de datos de resultados secuenciación del Laboratorio Nacional de Virología, Ministerio de Salud
Con la finalidad de investigar la asociación entre diferentes variantes del SARS-CoV-2 y la hospitalización se utilizó un modelo de regresión logística multivariada. Se observó que varias pre-variantes y variantes: "20A", "20B (Zeta)", "20C" y "Delta", se asociaron significativamente con un mayor riesgo de hospitalización, con ORs que oscilan entre 1.5 y 15. Estos hallazgos indican que las personas infectadas con estas variantes específicas del SARS-CoV-2 tuvieron una mayor probabilidad de requerir hospitalización en comparación con las que fueron infectadas con la pre-variante del clado 19B. Por otro lado, algunos resultados no mostraron significancia estadística, las variantes "20J (Gamma, V3)", "Omicron" y las variantes recombinantes no mostraron una asociación estadísticamente significativa con la hospitalización (figura 6)
En términos de la hospitalización, aproximadamente el 31.4% de los pacientes requirieron ser hospitalizados. Si bien este porcentaje no debe tomarse como una medida directa de la gravedad de la enfermedad, sugiere que una proporción significativa de pacientes presentó síntomas lo suficientemente graves como para requerir atención hospitalaria. Al determinar asociaciones, esta investigación reveló una asociación estadísticamente significativa entre las variantes 20A, 20B (Zeta), 20C y Delta y el riesgo de hospitalización con respecto a otras variantes.
Las variantes Delta y 20B (Zeta) también estuvieron asociadas a la probabilidad de infección en cuanto al aumento de la edad, esto sugiere que la edad pudo impulsar el riesgo de hospitalización cuando se combinaron con las características inherentes de estas variantes y las situaciones propias del brote, como la ausencia de inmunidad previa y vacunación, (en el caso de la pre-variante 20B, que circuló al inicio de la pandemia según GISAID). Por su parte, la variante Delta estuvo circulando desde Abril 2021 casi junto al inicio de la vacunación contra la COVID 19 en el país (Maier et al., 2022), a pesar de la inmunidad previa inducida por la exposición a otras variantes y a pesar de la vacunación, esta variante estuvo asociada a hospitalización, este mismo hecho fue reportado por diversos autores (Greene et al., 2023; Harris, 2022).
Es posible que las diferencias genéticas y características propias de las variantes y pre-variantes pudieran influir en su impacto clínico. La relación entre una variante específica y la hospitalización puede ser compleja y estar influenciada por varios factores que podrían incluir la combinación de ciertas mutaciones, la capacidad de transmisión, la respuesta inmunitaria del huésped, la edad, la efectividad de las medidas de control y prevención y las situaciones sociales que se vivían al momento de la circulación de cada variante o pre-variante.
Figura 6
Asociaciones entre la Hospitalización y las variantes identificadas en pacientes con COVID 19
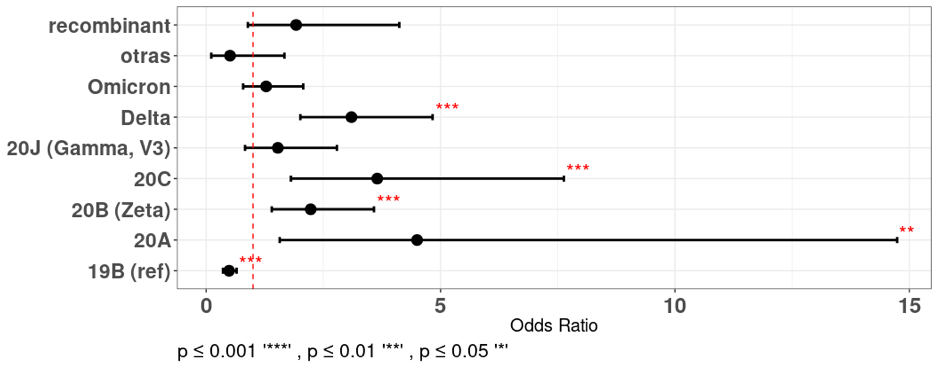
Fuente: Base de datos de resultados secuenciación del Laboratorio Nacional de Virología, Ministerio de Salud.
CONCLUSIONES
Este estudio permitió identificar y clasificar las variantes del SARS-CoV-2 mediante secuenciación genómica, destacando la presencia de mutaciones clave como S: D614G. Estos hallazgos son cruciales para entender la evolución y propagación del virus. Uno de los hallazgos más importantes es la relación entre la edad de los pacientes y la probabilidad de infección por diferentes variantes. Se observó que la variante 19B afectaba mayormente a pacientes jóvenes, mientras que las variantes Delta y 20B (Zeta) presentaron un riesgo mayor en individuos de mayor edad.
Además, el estudio evidenció que las variantes 20A, 20B (Zeta), 20C y Delta están asociadas a un aumento en el riesgo de hospitalización, con la edad jugando un rol significativo especialmente en las variantes 20B y Delta. Estos hallazgos subrayan la importancia de considerar factores demográficos en las investigaciones y gestión de brotes causados por la COVID-19, por lo tanto, aportan información valiosa para futuras investigaciones y estrategias de salud pública.
REFERENCIAS BIBLIOGRÁFICAS
Al-Rohaimi, A. H., & Al Otaibi, F. (2020). Novel SARS-CoV-2 outbreak and COVID19 disease; a systemic review on the global pandemic. Genes & Diseases, 7(4), 491–501. https://doi.org/10.1016/j.gendis.2020.06.004
Artic Network. (2020). Artic Network. https://artic.network/1-about.html
ARTIC SARS-CoV-2 Workflow. (2023). [Groovy]. EPI2ME Labs. https://github.com/epi2me-labs/wf-artic (Original work published 2021)
Brito, A. F., Semenova, E., Dudas, G., Hassler, G. W., Kalinich, C. C., Kraemer, M. U. G., Ho, J., Tegally, H., Githinji, G., Agoti, C. N., Matkin, L. E., Whittaker, C., Howden, B. P., Sintchenko, V., Zuckerman, N. S., Mor, O., Blankenship, H. M., de Oliveira, T., Lin, R. T. P., … Faria, N. R. (2022). Global disparities in SARS-CoV-2 genomic surveillance. Nature Communications, 13(1), Article 1. https://doi.org/10.1038/s41467-022-33713-y
Butt, A. A., Dargham, S. R., Chemaitelly, H., Al Khal, A., Tang, P., Hasan, M. R., Coyle, P. V., Thomas, A. G., Borham, A. M., Concepcion, E. G., Kaleeckal, A. H., Latif, A. N., Bertollini, R., Abou-Samra, A.-B., & Abu-Raddad, L. J. (2022). Severity of Illness in Persons Infected With the SARS-CoV-2 Delta Variant vs Beta Variant in Qatar. JAMA Internal Medicine, 182(2), 197–205. https://doi.org/10.1001/jamainternmed.2021.7949
Castañeda, S., Patiño, L. H., Muñoz, M., Ballesteros, N., Guerrero-Araya, E., Paredes-Sabja, D., Flórez, C., Gomez, S., Ramírez-Santana, C., Salguero, G., Gallo, J. E., Paniz-Mondolfi, A. E., & Ramírez, J. D. (2021). Evolution and Epidemic Spread of SARS-CoV-2 in Colombia: A Year into the Pandemic. Vaccines, 9(8), 837. https://doi.org/10.3390/vaccines9080837
El-Shabasy, R. M., Nayel, M. A., Taher, M. M., Abdelmonem, R., Shoueir, K. R., & Kenawy, E. R. (2022). Three waves changes, new variant strains, and vaccination effect against COVID-19 pandemic. International Journal of Biological Macromolecules, 204, 161–168. https://doi.org/10.1016/j.ijbiomac.2022.01.118
Farhud, D. D., & Mojahed, N. (2022). SARS-COV-2 Notable Mutations and Variants: A Review Article. Iranian Journal of Public Health, 51(7), 1494–1501. https://doi.org/10.18502/ijph.v51i7.10083
Freitas, A. R. R., Beckedorff, O. A., Cavalcanti, L. P. de G., Siqueira, A. M., Castro, D. B. de, Costa, C. F. da, Lemos, D. R. Q., & Barros, E. N. C. (2021). The emergence of novel SARS-CoV-2 variant P.1 in Amazonas (Brazil) was temporally associated with a change in the age and sex profile of COVID-19 mortality: A population based ecological study. The Lancet Regional Health - Americas, 1, 100021. https://doi.org/10.1016/j.lana.2021.100021
GISAID. (2022, April 4). GISAID - hCov19 Variants. hCov19 Variants. https://www.gisaid.org/hcov19-variants/
Greene, S. K., Levin-Rector, A., Kyaw, N. T. T., Luoma, E., Amin, H., McGibbon, E., Mathes, R. W., & Ahuja, S. D. (2023). Comparative hospitalization risk for SARS-CoV-2 Omicron and Delta variant infections, by variant predominance periods and patient-level sequencing results, New York City, August 2021–January 2022. Influenza and Other Respiratory Viruses, 17(1), e13062. https://doi.org/10.1111/irv.13062
Harris, J. E. (2022). COVID-19 Incidence and hospitalization during the delta surge were inversely related to vaccination coverage among the most populous U.S. Counties. Health Policy and Technology, 11(2), 100583. https://doi.org/10.1016/j.hlpt.2021.100583
Ihaka, R., & Gentleman, R. (1996). R: A Language for Data Analysis and Graphics. Journal of Computational and Graphical Statistics, 5(3), 299–314. https://doi.org/10.1080/10618600.1996.10474713
Korber, B., Fischer, W. M., Gnanakaran, S., Yoon, H., Theiler, J., Abfalterer, W., Hengartner, N., Giorgi, E. E., Bhattacharya, T., Foley, B., Hastie, K. M.,
Parker, M. D., Partridge, D. G., Evans, C. M., Freeman, T. M., de Silva, T. I., McDanal, C., Perez, L. G., Tang, H., … Montefiori, D. C. (2020). Tracking Changes in SARS-CoV-2 Spike: Evidence that D614G Increases Infectivity of the COVID-19 Virus. Cell, 182(4), 812-827.e19. https://doi.org/10.1016/j.cell.2020.06.043
Maier, H. E., Balmaseda, A., Saborio, S., Ojeda, S., Barilla, C., Sanchez, N., Lopez, R., Plazaola, M., Cerpas, C., van Bakel, H., Kubale, J., Harris, E., Kuan, G., & Gordon, A. (2022). Protection Associated with Previous SARS-CoV-2 Infection in Nicaragua. New England Journal of Medicine, 387(6), 568–570. https://doi.org/10.1056/NEJMc2203985
Maslo, C., Friedland, R., Toubkin, M., Laubscher, A., Akaloo, T., & Kama, B. (2022). Characteristics and Outcomes of Hospitalized Patients in South Africa During the COVID-19 Omicron Wave Compared With Previous Waves. JAMA, 327(6), 583–584. https://doi.org/10.1001/jama.2021.24868
Mishra, S., Mindermann, S., Sharma, M., Whittaker, C., Mellan, T. A., Wilton, T., Klapsa, D., Mate, R., Fritzsche, M., Zambon, M., Ahuja, J., Howes, A., Miscouridou, X., Nason, G. P., Ratmann, O., Semenova, E., Leech, G., Sandkühler, J. F., Rogers-Smith, C., … Flaxman, S. (2021). Changing composition of SARS-CoV-2 lineages and rise of Delta variant in England. EClinicalMedicine, 39, 101064. https://doi.org/10.1016/j.eclinm.2021.101064
Molina-Mora, J. A., Cordero-Laurent, E., Godínez, A., Calderón-Osorno, M., Brenes, H., Soto-Garita, C., Pérez-Corrales, C., Drexler, J. F., Moreira-Soto, A., Corrales-Aguilar, E., & Duarte-Martínez, F. (2021). SARS-CoV-2 genomic surveillance in Costa Rica: Evidence of a divergent population and an increased detection of a spike T1117I mutation. Infection, Genetics and Evolution, 92, 104872. https://doi.org/10.1016/j.meegid.2021.104872
Nanoporetech. (2022). Medaka [Python]. Oxford Nanopore Technologies. https://github.com/nanoporetech/medaka (Original work published 2017)
Nonaka, C. K. V., Gräf, T., Barcia, C. A. de L., Costa, V. F., de Oliveira, J. L., Passos, R. da H., Bastos, I. N., de Santana, M. C. B., Santos, I. M., de Sousa, K. A. F., Weber, T. G. L., Siqueira, I. C. de, Rocha, C. A. G., Mendes, A. V. A., & Souza, B. S. de F. (2021). SARS-CoV-2 variant of concern P.1 (Gamma) infection in young and middle-aged patients admitted to the intensive care units of a single hospital in Salvador, Northeast Brazil, February 2021. International Journal of Infectious Diseases, 111, 47–54. https://doi.org/10.1016/j.ijid.2021.08.003
Organización Mundial de la Salud. (2020, March 11). Alocución de apertura del Director General de la OMS en la rueda de prensa sobre la COVID-19 celebrada el 11 de marzo de 2020. https://www.who.int/es/director-general/speeches/detail/who-director-general-s-opening-remarks-at-the-media-briefing-on-covid-19---11-march-2020
Organización Panamericana de la Salud. (2022, December 28). Weekly COVID-19 Epidemiological Update -EW51- 28 December 2022—PAHO/WHO. PAHO. https://www.paho.org/en/documents/paho-weekly-covid-19-epidemiological-update-ew51-28-december-2022
Oxford Nanopore Technolgies. (2021). Oxford Nanopore launches ‘Midnight kit’, suitable for low to high-throughput SARS-CoV-2 sequencing, enabling rapid, low-cost, large-scale genomic surveillance of COVID-19. Oxford Nanopore Technologies. http://nanoporetech.com/about-us/news/oxford-nanopore-launches-midnight-kit-suitable-low-high-throughput-sars-cov-2
Prüβ, B. M. (2022). Variants of SARS CoV-2: Mutations, transmissibility, virulence, drug resistance, and antibody/vaccine sensitivity. Frontiers in Bioscience-Landmark, 27(2), Article 2. https://doi.org/10.31083/j.fbl2702065
Thulin, M. (2021). Chapter 8.3: Generalised linear models. In Modern Statistics with R (pp. 315–326). Eos Chasma Press.
Van Goethem, N., Chung, P. Y. J., Meurisse, M., Vandromme, M., De Mot, L., Brondeel, R., Stouten, V., Klamer, S., Cuypers, L., Braeye, T.,
Catteau, L., Nevejan, L., van Loenhout, J. A. F., & Blot, K. (2022). Clinical Severity of SARS-CoV-2 Omicron Variant Compared with Delta among Hospitalized COVID-19 Patients in Belgium during Autumn and Winter Season 2021–2022. Viruses, 14(6), Article 6. https://doi.org/10.3390/v14061297
Wick, R. (2022). Adapter trimmer for Oxford Nanopore reads [C++]. https://github.com/rrwick/Porechop (Original work published 2017)
Zhu, N., Zhang, D., Wang, W., Li, X., Yang, B., Song, J., Zhao, X., Huang, B., Shi, W., Lu, R., Niu, P., Zhan, F., Ma, X., Wang, D., Xu, W., Wu, G., Gao, G. F., & Tan, W. (2020). A Novel Coronavirus from Patients with Pneumonia in China, 2019. New England Journal of Medicine, 382(8), 727–733. https://doi.org/10.1056/NEJMoa2001017
© 2024 Revista Científica Estelí.
![]() Este trabajo está licenciado bajo una Licencia Internacional Creative Commons 4.0 Atribución-NoComercial-CompartirIgual.
Este trabajo está licenciado bajo una Licencia Internacional Creative Commons 4.0 Atribución-NoComercial-CompartirIgual.