
Enfermedad granulomatosa eosinofílica con poliangeítis: presentación de caso y revisión de la literatura
Eosinophilic granulomatous disease with polyangiitis: clinical case presentation and literature review.
Juan Enrique González Obando1, Miriam María Robles Cruz2, Dr. Douglas Rosales3
1
Estudiante del quinto año de Medicina, Facultad de Ciencias Médicas, UNAN-Managua.
2Médico Interno, Facultad de Ciencias Médicas, UNAN-Managua.
3Servicio de Hemato-oncología, Hospital Escuela Roberto Calderón Gutiérrez.
juengoob131196@gmail.com

RESUMEN
La enfermedad granulomatosa eosinofílica con poliangeítis es una vasculitis necrotizante sistémica que afecta a vasos de pequeño y mediano calibre. Es una enfermedad rara con una incidencia y prevalencia de 0,5-6 y 7-14 casos por millón de habitantes por año, respectivamente.
La etiología continúa sin conocerse; se han involucrado factores genéticos y ambientales y hay una relación con anticuerpos ANCA y anti-MPO. La enfermedad evoluciona en tres fases sucesivas: el primer estadio se caracteriza por pródromos, con manifestaciones alérgicas como rinitis, asma, pólipos nasales, entre otras; un segundo estadio donde aparece hipereosinofilia sanguínea e infiltrados tisulares de eosinófilos; y el tercer estadio donde se presenta vasculitis sistémica que afecta principalmente piel, sistema nervioso periférico y riñones. El diagnóstico se realiza sobre la base clínica, hallazgos de laboratorio (eosinofilia y otros), anticuerpos e histopatología; el pronóstico es bueno, pero hay alta morbilidad, el riesgo de recidivas es del 35%. El tratamiento debe ser individualizado y se realiza con corticoides y ciclofosfamida.
Se presenta el caso de paciente masculino de 56 años de edad, con antecedentes de rinitis alérgica y alergia a betalactámicos, que acude a consulta por fiebre prolongada. Se le realizan exámenes de rutina y se aprecia hipereosinofilia, posterior a su ingreso presenta neuropatía periférica en miembros superiores e inferiores; se le efectúa estudios de extensión, encontrándose ANCA positivos y una biopsia de médula ósea mostrando predominio de gránulos eosinófilos; fue tratado con corticoides orales y pregabalina con mejoría evidente, a los nueve meses de seguimiento se encuentra en remisión, siendo valorado mensualmente.
Palabras claves: enfermedad granulomatosa eosinofílica con poliangeítis, anticuerpos anticitoplasma de neutrófilos, hipereosinofilia.
ABSTRACT
Eosinophilic granulomatosis with polyangitis is a systemic necrotizing vasculitis, which involves small and medium sized vessels. It is a rare disease with and incidence and prevalence of 0.5-6 and 7-14 cases per million of habitants per year, respectively.
Until now, the etiology remains unknown; genetic and environmental factors have been involved, along with the ANCA and Anti MPO antibodies. The disease evolves in three successive phases: the first stage is characterized by prodromes, with allergic manifestations such as rhinitis, asthma, nasal polyps, among others; a second stage where blood hypereosinophilia and eosinophil tissue infiltrates appear; and the third stage where systemic vasculitis occurs mainly affecting the skin, peripheral nervous system and kidneys. The diagnosis is made on the clinical basis, laboratory findings (eosinophilia and others), antibodies and histopathology. The prognosis is good, but there is high morbidity, the risk of recurrence is 35%. The treatment must be individualized and performed with corticosteroids and cyclophosphamide.
We present the case of a 56 years old male patient with history of allergic rhinitis and allergy to betalactamics who develops prolonged fever. Routine lab tests report hyper eosinophilia, later on he develops peripheric neuropathy in upper and lower limbs. Further studies result positive for ANCA and a bone marrow biopsy shows predominance of eosinophilic granules. This patient was treated with oral corticosteroids and pregabalin with evident improvement, nine months after treatment he is in remission and being followed every month.
Key Words: Eosinophilic granulomatosis with polyangitis, antineutrophil cytoplasmic antibody, Hypereosinophilia
INTRODUCCIÓN
La enfermedad granulomatosa eosinofílica con poliangeitis (EGPA), también conocido con los nombres de granulomatosis alérgica o angeítis granulomatosa alérgica, o síndrome de Churg Strauss es una vasculitis necrotizante sistémica que afecta a vasos de pequeño calibre y en menor proporción, de mediano calibre1.
Fue descrito desde el punto de vista histopatológico por primera vez, en 1951, por Jacob Churg (1910-2005) y Lotte Strauss (1913-1985), anatomopatólogos del hospital Mount Sinai de New York.

El American College of Rheumatology (ACR) en 1990, definió la enfermedad mediante sus criterios de clasificación (tabla 1) que son los más aceptados en la actualidad. La presencia de al menos cuatro de los seis criterios confirió una sensibilidad y especificidad diagnósticas del 85% y 99,7% respectivamente, en la población estudiada1.Finalmente, la EGPA se ha clasificado junto con la granulomatosis de Wegener (GW) y la poliangeítis microscópica (PAM) como una de las vasculitis asociadas a anticuerpos antineutrófilicos citoplasmáticos (ANCA)1.
REPORTE DEL CASO
Hombre de 56 años de edad, originario de Carazo quien previo a su ingreso presentó fiebre de un mes de duración, intermitente, con episodios cada 12 horas, aproximadamente, que oscilaba entre los 38.5 a 39.5 grados Celsius. En el mismo mes, refiere anorexia y debilidad en miembros inferiores, por lo que acude a consulta privada, donde se realiza biometría hemática completa (BHC) que reporta hipereosinofilia, razón por la cual se solicita referencia al servicio de Hemato-Oncología del Hospital Roberto Calderón Gutiérrez.
Dentro de los antecedentes patológicos de relevancia, se aprecia rinitis alérgica, de inicio en la niñez, y alergia a fármacos betalactámicos. Antecedentes personales no patológicos negados.
Al examen físico, al ingreso los signos vitales se encontraron en rangos normales, a excepción de la temperatura, que fue de 39.4 °C, a nivel de piel y mucosas con palidez mucocutánea, sin otros hallazgos de relevancia. La revisión de cabeza, cuello, tórax y abdomen se encontró en parámetros normales, a la exploración neurológica de los miembros superiores se encontró disminuida la sensibilidad a la presión y a la vibración, en la exploración de los miembros inferiores la fuerza muscular fue dos de cinco.
Posterior al ingreso, se realiza una BHC donde se destaca una hipereosinofilia de 3,544/ uL y anemia normocítica normocrómica, con hemoglobina de 11,5 g/dl. Después de su ingreso se le realizan BHC de rutina las que en todo momento reportaron hipereosinofilia, siendo el valor más elevado de 6,869/uL, al igual que anemia normocítica normocrómica. Debido a lo anterior se sospecha de una parasitosis tisular, por lo que se administra metronidazol 400 mg vía intravenosa cada 12 horas por cinco días sin resultado positivo, y luego albenzadol 400 mg vía oral y de igual manera no se reporta mejoría.

Se elaboró una serie de estudios diagnósticos de laboratorio, siendo resultados normales (tabla 2).
Se realizó endoscopía digestiva alta que reportó candidiasis esofágica y gastropatía crónica, causada por Helicobacter pylori. También se realiza rectosigmoidoscopía, mostrando hemorroides internas grado II. Así mismo, se indican radiografía postero anterior de tórax, ecocardiograma y ultrasonido abdominal, donde no se encuentran hallazgos patológicos. Se realizó biopsia de médula ósea (figura 1) en la cual se observó hipocelularidad para la edad del paciente, con abundante hemorragia, se observaban las tres series mieloide y eritroide en sus diversos estados de maduración con una relación 3 a 1, megacariocitos presentes. Las células mieloide muestran en un 50% gránulos eosinofílicos.
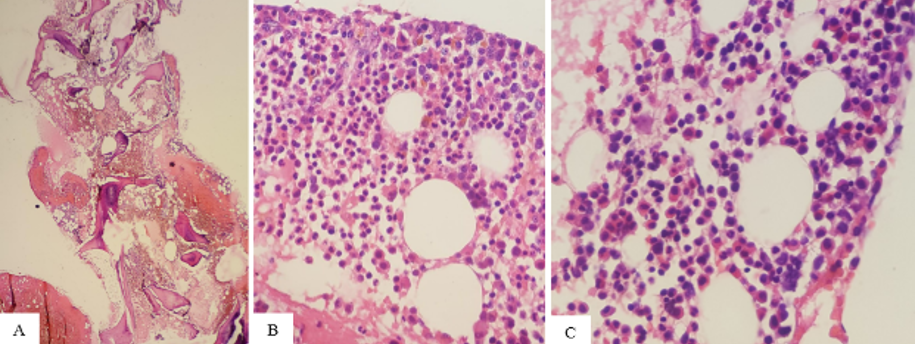
Figura 1: A. HE.4X, Médula ósea hipocelular para la edad, abundante material hemático. B y C. HE.40X, Serie granulocítica con predominio de diferenciación eosinofílica.
Cortesía del Departamento de Patología Hospital Roberto Calderón.
Cuatro meses posteriores al ingreso, se refiere al paciente a la consulta privada de hematología a la ciudad de México, para realizar estudios de extensión allá disponibles. Durante su evolución el paciente presentó desnutrición proteico calórica, con un IMC de 17.7 y albúmina de 2.8 g/dl. Para el tratamiento de la neuropatía periférica se administra pregabalina, 150 mg diarios y, posteriormente, se aumenta la dosis a 300 mg asociados a fisioterapia.
Se realiza estudios de citogenética convencional, que mostraron un resultado de 46 XY, siendo normal para su género, de igual manera se descarta procesos mieloproliferativos a través de la citometría de flujo. Se descartan procesos mieloproliferativos asociados a rearreglos de PDGFR alfa, PDGFR beta y mutación del FLT3; del mismo modo, se realiza una electroforesis policlonal, detectándose hiperglobulinemia, con predominio de IgG e IgA, los valores de IgM eran normales. Se le realiza tomografía de tórax y abdomen, descartándose neoplasias. Se realiza ANCA, siendo positivos, con títulos de 1:40.
Con la presencia de neuropatía periférica, hipereosinofilia y ANCA positivo, se realiza el diagnóstico de enfermedad granulomatosa eosinofílica con poliangeítis o síndrome de Churg Strauss, y se inicia manejo con prednisona a dosis de 75 mg diarios por vía oral.
Cuando el paciente regresa a Nicaragua se realiza, en el Hospital Roberto Calderón, una biometría hemática de control, presentando eosinófilos 420/uL, hemoglobina 12.2 g/dl. Se inicia la disminución paulatina mensual de la dosis de prednisona.
A los cinco meses de seguimiento, el paciente presenta mejoría clínica, según biometrías hemáticas de control que se han encontrado en parámetros normales. Actualmente, se encuentra en período de remisión, y en seguimiento mensual por consulta externa de hematología.
Comentarios adicionales al caso
La enfermedad granulomatosa eosinofílica con poliangeítis es una patología de variadas manifestaciones clínicas. Una manera de clasificarla es en dependencia del examen serológico ANCA que puede ser ANCA positivo o ANCA negativo. En el caso del paciente descrito anteriormente, su sintomatología era del fenotipo clínico ANCA positivo.
Consideraciones éticas
Mediante la firma del consentimiento informado del paciente se recibió autorización de parte del servicio de Hemato-Oncología del Hospital Escuela Dr. Roberto Calderón Gutiérrez para realizar la publicación del caso, además, se solicitó el permiso del departamento de patología del hospital para el uso de la biopsia del paciente.
DISCUSIÓN
La enfermedad granulomatosa eosinofílica con poliangeítis es una enfermedad rara con una incidencia aproximada de 0,5-6 casos por millón de habitantes / año y una prevalencia de 7-14 casos por millón. Suele aparecer entre la tercera y quinta década de la vida (edad media 45-50 años, rango 2-85 años). Su presentación es infrecuente en mayores de 65 años y en niños1, no predomina en ningún género.
Se ha descrito la evolución de la enfermedad en tres fases sucesivas: un primer estadio que se caracteriza por pródromos donde principalmente aparecen manifestaciones alérgicas como rinitis, asma, pólipos nasales, alergias a fármacos, a alimentos, al polvo o polen, un segundo estadio aparece la hipereosinofilia sanguínea e infiltrados tisulares de eosinófilos: infiltración miocárdica, neumonía eosinofílica de Löffler y gastroenteritis eosinofílica. Finalmente, el tercer estadio consiste en el desarrollo de vasculitis sistémica con afectación de diversos órganos, principalmente piel, sistema nervioso periférico y riñones1. Aunque estas tres fases no necesariamente deben ser consecutivas, las series de casos más extensas y completas han descrito las características clínicas y serológicas de sus pacientes en el contexto de estas fases3. En el caso antes descrito se trata de un paciente que puede clasificarse dentro del segundo estadio.
Se ha postulado la existencia de dos fenotipos de la enfermedad. Se sabe que los pacientes con ANCA positivos tienen en mayor frecuencia la presencia de manifestaciones constitucionales, sinusitis, glomerulonefritis, hemorragia alveolar, púrpura cutánea, mononeuritis múltiple, afectación de sistema nervioso central y la presencia de vasculitis en las muestras de biopsias. Por otro lado, en los ANCA negativos ocurre con mayor frecuencia la fiebre, pericarditis, miocardiopatía, infiltrados pulmonares, derrame pleural y livedo reticularis2. En el caso de nuestro paciente se encontraron ANCA positivos, asociados con las manifestaciones constitucionales, síntomas alérgicos y desarrollo de neuropatía periférica, todo ello en correspondencia con lo descrito en la literatura.
La presentación clínica típica de la EGPA es la aparición de manifestaciones vasculíticas en un paciente con historia de rinitis alérgica, poliposis nasal y asma de inicio tardío, usualmente preexistente por 5 a 10 años. Los síntomas generales, como fiebre o pérdida de peso, mononeurítis múltiple y/o manifestaciones cutáneas como púrpura son las más frecuentes al inicio de la enfermedad, asociadas al recuento de eosinófilos elevado y síndrome inflamatorio4. Las artralgias y mialgias generalizadas han sido reportadas en un 3%7 al 57% de casos3.
El asma parece ser la característica común de todos los pacientes con EGPA, ya que aparece del 96% al 100% de pacientes y es la característica principal de la fase prodrómica. La latencia típica entre la aparición del asma y la fase vasculítica se estima en un promedio de 3 a 9 años. En su instauración el asma es generalmente severa, poco controlable y refractaria a la medicación inhalada, se asocia con antecedentes de rinitis alérgica en 47% al 93% de casos y de sinusitis paranasal crónica y recurrente en 62%-77%3. En el caso presentado, el cuadro clínico se centró principalmente en manifestaciones alérgicas en forma de rinitis crónica y el desarrollo de neuropatía periférica, sin embargo, no se apreciaron signos clínicos ni radiológicos sugestivos de compromiso pulmonar, probablemente porque se realizó captación del mismo en su fase más temprana.
Los hallazgos de laboratorio anormales incluyen anemia, leucocitosis, PCR y velocidad de sedimentación globular elevada4. La eosinofilia en sangre periférica (>10% en el conteo diferencial de glóbulos blancos o >1,5 x 109/L) es probablemente lo más sobresaliente de la EGPA y puede caracterizar cualquier fase de la enfermedad3. Puede haber una amplia gama de cambios rápidos en el recuento de eosinófilos4.
Los ANCA están presentes en más de la mitad de los pacientes con un patrón de tinción perinuclear. La positividad para ANCA debe ser confirmada por demostración de anticuerpos antimieloperoxidasa (MPO) en suero4. En el caso antes detallado se presentaron las alteraciones de laboratorio descritas en la literatura, siendo todas ellas inespecíficas a excepción de la positividad de los ANCA.
Antes del inicio de la vasculitis, la eosinofilia concomitante, asma y los infiltrados pulmonares pueden semejar ciertas infecciones parasitarias, como helmintiasis, o aspergilosis broncopulmonar alérgica5.
Dentro de los diagnósticos diferenciales se encuentra el síndrome hipereosinofílico (SHE)6. Esta es una condición crónica caracterizada por una eosinofilia en sangre periférica sostenida y persistente por más de seis meses consecutivos. Los signos de vasculitis y ANCA están ausentes en el SHE3.
En lo que respecta a otras vasculitis sistémicas es ampliamente conocido que la granulomatosis de wegener (GW) y la poliangeítis microscópica (PAM) pueden afectar los mismos órganos que la EGPA y también pueden ser ANCA positivos, pero la presencia de asma y eosinofilia es poco común; por otro lado, el involucro renal en estas entidades suele ser la característica principal, algo que no sucede en EGPA3.
En el caso descrito se inició el abordaje de la hipereosinofilia asociada a fiebre y pruebas de laboratorio que orientaban a un proceso inflamatorio sistémico como un SHE secundario a una parasitosis tisular, por lo que se decidió administrar antiparasitarios sistémicos, sin cambio alguno. Se descartó la posibilidad de antecedentes atópicos. Se eliminaron de igual forma otras causas de SHE secundario, principalmente respiratorias, gastrointestinales y reumáticas. Posteriormente se planteó la posibilidad de un SHE primario por lo que se realizaron estudios de extensión para descartar trastornos mieloproliferativos y neoplasias, sin datos concluyentes. En México, se realizan estudios especializados que permiten descartar todas las causas antes mencionadas y se encuentran ANCA positivos con lo cual se llega al diagnóstico, interpretando todo lo anterior en el contexto clínico.
El pronóstico en general es bueno, pero con alta morbilidad. Recidiva una o más veces el 35% de los casos. Incluso en los casos de buena evolución puede permanecer el asma con sintomatología grave, secuelas de la neuropatía, alteraciones crónicas cardíacas, renales, del sistema nervioso central u oftalmológicas1-9.
El five factor score de Guillevin ha sido de utilidad para establecer un pronóstico. Se trata de un sistema de valoración pronostica basado en cinco factores de riesgo: 1) insuficiencia renal (niveles de creatinina >1,6 mg/dl ó 141 mmol/L); 2) proteinuria >1 gramo/24 horas; 3) sangrado, perforación, infarto gastrointestinal o pancreatitis; 4) afectación del SNC y 5) cardiomiopatía3.
En la actualidad se cuenta con una amplia gama de medidas terapéuticas, el cual se tendrá que individualizar en cada caso dependiendo de la gravedad de la enfermedad y de las características del paciente1. Como tratamiento de inducción se tiene corticoesteroides como prednisona o metilprednisolona, y en algunos casos se utiliza ciclofosfamida oral o intravenoso7.
CONCLUSIONES
La enfermedad granulomatosa eosinofílica con poliangeítis es una vasculitis necrotizante sistémica que afecta a vasos de pequeño calibre. Es una enfermedad rara con una incidencia y prevalencia de 0.5-6 y 7-14 casos por millón de habitantes/año, respectivamente; aparece entre los 30 y 50 años y no tiene predilección por sexo. Se desarrolla en tres fases: una prodrómica, una segunda fase con hipereosinofilia sanguínea e infiltrados tisulares de eosinófilos y finalmente una fase vasculítica sistémica. Tiende a debutar con manifestaciones alérgicas como rinitis, poliposis nasal y asma de inicio tardío y difícil manejo. El diagnóstico se realiza usualmente a través de la biopsia de las lesiones ubicadas principalmente en piel, por la positividad de los ANCA con patrón perinuclear y la hipereosinofilia sanguínea, asociados al contexto clínico. El tratamiento se realiza con corticoides y ciclofosfamida. El pronóstico es bueno, sin embargo, se presentan recidivas en 35% de casos. Dado que se trata de una enfermedad infrecuente, sus manifestaciones pueden pasar desapercibidas y llevar a un diagnóstico tardío, permitiendo el avance silencioso de la misma. Por ello, se insta a incluir dentro del pensamiento médico las características clínicas y de laboratorio antes descritas con el fin de realizar un buen abordaje.
RECOMENDACIONES
Para realizar el diagnóstico de EGPA se requiere del uso de diversos estudios de extensión no disponibles en países de bajos recursos que permiten principalmente hacer el diagnóstico diferencial; entre los que se pueden mencionar la citogenética convencional, citometría de flujo, con especial énfasis en las mutaciones de los genes PDGFR alfa y beta, así como del gen FLT3. Por lo tanto, se enfatiza en que, a pesar de lo infrecuente de su ocurrencia, la enfermedad tiene una forma característica de presentación bien descrita en la literatura y que debe originar la sospecha clínica en los médicos tratantes.
REFERENCIAS BIBLIOGRÁFICAS
- Castellano Cuesta J, González Domínguez, J, Fernández-Llanio Comella, N. Síndrome de Churg-Strauss. Enfermedades reumáticas: Actualización SVR. [Internet]. 2013[Consultado 5 Jul 2017]:277-88. Recuperado a partir de: http://www.svreumatologia.com/wp-content/uploads/2008/04/Cap-15-Sindrome-de-Churg-Strauss.pdf
- Greco A, Rizzo MI, De Virgilio A, Gallo A, Fusconi M, Ruoppolo G, et al. Churg–Strauss syndrome. Autoimmunity Reviews. [Internet]. 2015[Consultado 5 Jul 2017];14(4):341-8. doi:10.1016/j.autrev.2014.12.004
- Baldini C, Talarico R, Della Rossa A, Bombardieri S. Clinical manifestations and treatment of Churg-Strauss syndrome. Rheumatic diseases clinics of North America. [Internet]. 2010[Consultado 25 Jul 2017];36(3):527-43. doi: 10.1016/j.rdc.2010.05.003.
- Rolla G, Boita, M, Heffler, E, Guida, G. Churg Strauss Syndrome: Clinical and Pathogenetic Approach to Therapy. 2011[Consultado 20 Jul 2017]. En: Advances in the Diagnosis and Treatment of Vasculitis [Internet]. InTech; [235-52]. Recuperado a patir de: http://www.intechopen.com/books/advances-in-the-diagnosis-and-treatment-of-vasculitis/churg-strausssyndrome-clinical-and-pathogenetic-approach-to-therapy
- Mouthon L, Dunogue B, Guillevin L. Diagnosis and classification of eosinophilic granulomatosis with polyangiitis (formerly named Churg–Strauss syndrome). Journal of Autoimmunity. [Internet]. 2014 [Consultado 5 Sept 2017];48-49:99-103. doi:10.1016/j.jaut.2014.01.018
- Narula N, Narula, T., Derbes, S., Espinoza, L. Churg-Strauss Angiitis. The American Journal of the Medical Science. [Internet]. 2014[Consultado 3 Agosto 2017];348(6):522-7. doi: 10.1097/MAJ.0b013e31829f8306
- Ramentol-Sintas M, Martínez-Valle F, Solans-Laqué R. Churg–Strauss Syndrome: An evolving paradigm. Autoimmunity Reviews. [Internet]. 2012[Consultado 10 Agosto 2017];12(2):23 doi:10.1016/j.autrev.2012.07.009
- Mahr A, Moosig F, Neumann T, Szczeklik W, Taille C, Vaglio A, et al. Eosinophilic granulomatosis with polyangiitis (Churg-Strauss): evolutions in classification, etiopathogenesis, assessment and management. Current opinion in rheumatology. [Internet] 2014[Consultado 20 Agosto 2017];26(1):16-23. doi: 10.1097/BOR.0000000000000015
- Vaglio A, Moosig F, Zwerina J. Churg-Strauss syndrome: update on pathophysiology and treatment. Current opinion in rheumatology. [Internet] 2012[Consultado 5 Oct 2017];24(1):24-30 doi: 10.1097/BOR.0b013e32834d85ce.