
Gerardo Mejía-Baltodano1, Alba Nubia Montenegro2, María Lisseth Vallejos3, Alejandra Jirón 4,
Nubia Talavera5, Violeta Alemán6
1Registro Nicaragüense de Anomalías Congénitas, Departamento de Genética Ministerio de Salud, Nicaragua; mejiabaltodano@gmail.com, https://orcid.org/0000-0002-1696-8310
2Hospital Alfonso Moncada, Ocotal, Nueva Segovia, Nicaragua; albanubia_montenegro@yahoo.com; https://orcid.org/0009-0000-0709-2831
3 Servicio de Neonatología, Hospital Infantil “Manuel de Jesús Rivera”, Managua, Nicaragua; malivallejos@hotmail.com; https://orcid.org/0009-0007-6566-4106
4Servicio de Neonatología, Hospital Bertha Calderón, Managua, Nicaragua; andreitajironb@gmail.com; https://orcid.org/0000-0002-1882-3094
5Servicio de Neonatología, Hospital Gaspar García Laviana, Rivas, Nicaragua; nubiatalavera2011@hotmail.com; https://orcid.org/0009-0004-6253-9842
6Departamento de Cirugía, Hospital Infantil “Manuel de Jesús Rivera”, Managua, Nicaragua; violeta.aleman@yahoo.fr; https://orcid.org/0009-0002-9773-9532
Autor por correspondencia: Dr. Gerardo Mejía-Baltodano mejiabaltodano@gmail.com

El síndrome de Fraser es un síndrome genético con herencia autosómica recesiva. Se presenta con diversas anomalías congénitas, pero de manera preponderante incluye la presencia de criptoftalmos uni o bilateral, sindactilia cutánea y anomalías genitourinarias. Una serie de anomalías en otros órganos y sistemas han sido reportados. Presentamos el caso de una paciente con diagnóstico clínico de Síndrome de Fraser con diversas anomalías congénitas, incluyendo algunas que han sido reportadas muy pocas veces en la literatura internacional, se trata además del primer caso de Síndrome de Fraser reportado en Nicaragua.
Palabras clave: Síndrome de Fraser, Criptoftalmos, persistencia de cloaca
Fraser síndrome is a genetic syndrome with autosomal recessive inheritance. It present with many congenital anomalies, but predominantly includes the presence of uni or bilateral cryptophthalmos, cutaneous syndactyly and genitourinary anomalies. A number of abnormalities in other organs and systems have been reported. We present a patient with a clinical diagnosis of Fraser syndrome with many congenital anomalies, including some that have been reported very rarely in the international literature, it is also the first case of Fraser syndrome reported in Nicaragua.
Key words: Fraser syndrome, Cryptophthalmos, persistent cloaca
El síndrome de Fraser es un síndrome genético autosómico recesivo muy poco frecuente, también conocido como síndrome de Criptoftalmos-Sindactilia o Criptoftalmos con otras malformaciones, síndrome de Fraser-Francoise, síndrome de Meyer-Schwickerath o síndrome de Ullrich-Feichtiger 1, se caracteriza básicamente por la presencia de criptoftalmos, sindactilia y anomalías en órganos genitales; sin embargo, otras anomalías congénitas en diferentes estructuras corporales han sido reportadas por otros autores. En 2007, Van Haelst et al.2 hicieron una revisión de los criterios mayores y menores que habían sido establecidos por Thomas et al.3, por lo que propusieron una modificación a los criterios diagnósticos, y consideran que es posible establecer el diagnóstico basados en la presencia de 3 criterios mayores, o 2 criterios mayores y 2 criterios menores, o 1 criterio mayor y 3 criterios menores. Su incidencia se considera menor de 0.043 por 10,000 nacidos vivos y 1.1 en 10,000 fetos4. Según EUROCAT la prevalencia en Europa es de 0.2 por 10,000 nacidos vivos basado en su registro de 12, 886,464 nacimientos5. En la base de datos OMIM, se reportan 3 entradas para síndrome de Fraser, llamadas síndrome de Fraser 1, 2 y 3, que presentan un espectro de anomalías congénitas muy similar, pero que son causadas por mutaciones en diferentes genes; las tres con herencia autosómica recesiva. Fraser 1 por mutaciones en el gen FRAS1 ubicado en el cromosoma 4q21.21; Fraser 2 por mutaciones en el gen FREM2 en el cromosoma 13q13.3 y Fraser 3 por mutaciones en el gen GRIP1 en el cromosoma 12q14.3 6.
Este primer caso clínicamente diagnosticado con Síndrome de Fraser, ha sido reportado por el sistema de vigilancia de anomalías congénitas en Nicaragua, que, como hallazgo muy pocas veces reportado, presentó anomalía tipo persistencia de cloaca además de otras anomalías ya antes reportadas en este síndrome.
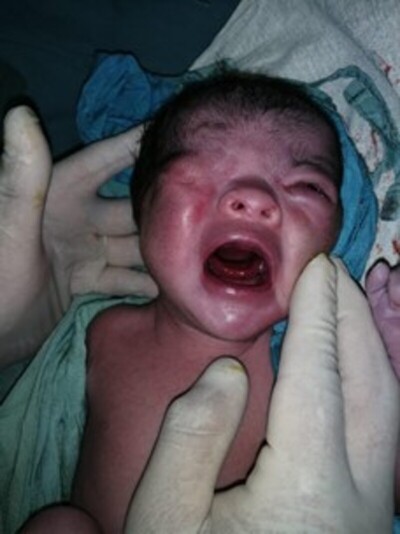
Paciente producto del séptimo embarazo de madre de 36 años, con un aborto previo y 5 hijos sanos, padre de 33 años, no consanguíneos, sin antecedentes de importancia personales ni familiares. Nace vía vaginal en un hospital primario del norte de Nicaragua, con historia de haberse realizado 8 controles prenatales y ultrasonidos obstétricos a las 28 y 36 semanas de gestación reportados normales. Nace a las 37 semanas y 5 días de gestación, con peso de 2,750 gramos, talla de 48 cms, perímetro cefálico de 34 cms, con APGAR 8/9. Al examen físico se observa oclusión total de ojo derecho (fig.1), no presenta datos de descompensación cardiopulmonar, no se logra diferenciar adecuadamente el sexo y se observa que presenta ano imperforado por lo que se decide su traslado a hospital departamental.
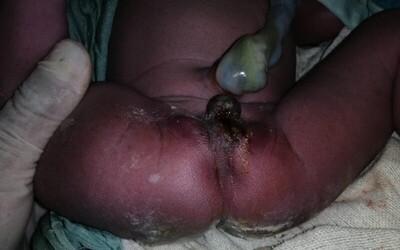
En hospital departamental observaron que tiene una fístula hacia la región perineal donde se evidencia salida de orina y meconio, por lo que se considera la posibilidad de malformación ano rectal tipo cloaca (fig. 2), además observan que presenta sindactilia cutánea entre II y III dedos y entre III y IV dedos en ambas manos; se decide traslado a Hospital Pediátrico de Referencia Nacional Manuel de Jesús Rivera en la capital del país, ya que se considera necesario valorar por cirugía pediátrica. Es recibido en condiciones estables, sin dificultad respiratoria, con presencia de estridor laríngeo al llanto, sospechando laringomalacia o estenosis laríngea que no compromete la mecanica ventilatoria por lo que no amerita ningún procedimiento. Se procede a realizar diversas evaluaciones especializadas y exámenes de rutina, encontrando BHC, Bilirrubinas, Na, K, Cl, Ca, Urea, Nitrógeno de urea, Creatinina, TP, TPT e INR normales, el estudio de PCR fue negativo.
Entre los estudios de imagen se realizó ultrasonido transfontanelar reportado normal y ultrasonido abdominal que reveló agenesia renal derecha. La valoración cardíaca reportó presencia de Comunicación Inter Auricular (CIA) tipo ostium secundum con Persistencia de Conducto Arterioso (PCA). Se realizó ultrasonido ocular considerando que el ojo izquierdo es normal, mientras que en el derecho se reporta que solo se logra ver el cristalino engrosado; sin embargo, se sugiere realizar tomografía axial de ojo derecho. La valoración por cirugía pediátrica confirmó la sospecha de anomalía tipo cloaca y se realizó cirugía de colostomía tipo Peña; durante el procedimiento quirúrgico se evidencia la presencia de trompas de Falopio, ovarios y útero. La valoración genética además de todo lo antes descrito encuentra la presencia de microtia bilateral grado II con pabellones auriculares de pequeño tamaño con conductos auditivos estenóticos. A nivel genital hay falo de unos 2.5 centímetros de longitud con fusión labio-escrotal y se visualiza fístula hacia perineo por donde se evidencia salida de material fecal y orina por lo que se considera malformación ano-rectal tipo cloaca y se considera la posibilidad de Síndrome de Fraser. La valoración oftalmológica reporta en ojo izquierdo la presencia de coloboma de párpado superior y no fue posible evaluar el ojo derecho por no poder realizar apertura palpebral, pero se describe que se palpa globo aumentado de tamaño respecto al izquierdo. En una nueva valoración oftalmológica se realiza otro ultrasonido ocular reportándose que no es posible visualizar las estructuras de la cámara anterior del globo ocular derecho, se recomienda seguimiento en 6 meses. La evolución fue satisfactoria después de 16 días de hospitalización por lo que se decidió egreso hospitalario con cita a varias especialidades médicas.
Según los criterios diagnósticos establecidos por Thomas et al. 3 y revisados por van Haelst et al. 2, nuestro paciente reúne 5 de los 6 criterios mayores y 2 de los 5 criterios menores reportados en el síndrome de Fraser: criptoftalmos, sindactilia, anomalías en genitales, anomalías del sistema urinario y anomalías laríngeas o traqueales, faltando únicamente el criterio de tener un hermano igualmente afectado. Con respecto a otras anomalías asociadas a los criterios mayores considerados como criterios menores, nuestro paciente presenta anomalías anorectales (malformación anorectal tipo cloaca) y de pabellones auriculares (microtia bilateral grado II con conductos auditivos externos estenóticos), además de ano imperforado con fístula perineal con salida de meconio y orina (malformación ano-rectal tipo cloaca) y cardiopatía congénita tipo CIA ostium secundum con persistencia de conducto arterioso (PCA).
Con relación al criptoftalmos en nuestro paciente fue unilateral derecho. En el ojo izquierdo, solamente se encontró coloboma de párpado superior. Es importante mencionar que en un estudio realizado en España por Martínez-Frías et al 7 de 7 casos, 4 de ellos tenían criptoftalmos, es decir, dicha anomalía no siempre está presente, aunque es un dato clínico que llama mucho la atención hacia la posibilidad de diagnosticarlo. Los reportes de criptoftalmos unilateral en síndrome de Fraser son pocos, ya que la mayoría de las veces se presenta bilateral, sin embargo, en una revisión de casos publicados entre 1986 y 2002, Slavotinek y Tifft 8, reportan criptoftalmos en 103 de 117 pacientes siendo en 32 casos (27.4%) unilateral.
Otro hallazgo importante de mencionar en nuestro paciente fue la malformación ano-rectal (MAR) tipo cloaca con la dificultad de poder asignar el sexo desde el nacimiento ya que se observó un falo y fusión de los pliegues labio-escrotales, sin embargo, durante el procedimiento quirúrgico se observan internamente estructuras femeninas (útero, trompas y ovarios) y ausencia de estructuras internas masculinas. Los reportes de cloaca persistente en síndrome de Fraser existen, pero son muy raros. En el estudio de Martínez-Frías et al7 con datos del ECEMC (Estudio Colaborativo Español de Malformaciones Congénitas) entre abril 1976 y marzo 1997 con un total de 1,405,374 recién nacidos vivos y 9,042 recién nacidos fallecidos, reportan 7 casos de síndrome de Fraser y solo 1 de ellos con persistencia de cloaca.
Las anomalías cardíacas son pocas veces reportada en síndrome de Fraser y nuestra paciente tenía CIA tipo osteum secundum y PCA. En una revisión realizada por EUROCAT 9 entre enero de 1990 y diciembre de 2008, identificaron 26 casos de síndrome de Fraser en una población de 12,886,464 nacimientos y solamente 3 de los 26 presentaron problemas cardíacos. Al realizarse una búsqueda en PubMed usando las palabras “Crypthophtalmos” y “Fraser síndrome”, encontramos 301 artículos publicados entre 1952 y 2019 en los cuales fue evidente la poca cantidad de publicaciones relacionadas con problemas cardíacos y gastrointestinales. Nuestro paciente presentó defecto cardíaco tipo CIA-OS (comunicación interauricular ostium secundum), una alteración no reportada en la revisión de Pub-Med y que además en el OMIM no se registra como un hallazgo en ninguno de las tres entradas acerca de síndrome de Fraser, sin embargo, han sido reportados casos de síndrome de Fraser con cardiopatías congénitas10, 11, este tipo de anomalía congénita no es reconocido como parte del espectro de anomalías en ninguno de los tres tipos de síndrome de Fraser que han sido descritos en OMIM.6
Nuestro paciente fue intervenido quirúrgicamente por presentar ano imperforado con fístula recto-uretro-vaginal lo que hizo sospechar anomalía tipo persistencia de cloaca, comprobado transquirúrgicamente. Por otro lado, las alteraciones gastrointestinales también han sido reportadas, aunque de igual manera en pocas publicaciones, como por ejemplo la publicación de Narang M. et al. 12, reporta un paciente con atresia colónica. Alvarés-Neri et al. 13 describe en su estudio, a una recién nacida con alteración tipo cloaca que además presentó anomalías laríngeas, sindactilia y criptoftalmos.
Ante la ausencia en nuestro país de un laboratorio de genética, para poder realizar estudios moleculares para enfermedades genéticas, que nos permita contundentemente demostrar el diagnóstico, consideramos que nuestra paciente presenta suficientes características clínicas para soportar el diagnóstico clínico de Síndrome de Fraser, primer caso reportado a través del sistema de vigilancia del Registro Nicaragüense de Anomalías Congénitas.
El espectro de anomalías congénitas presentado por nuestra paciente se corresponde clínicamente con el observado y reportado en el síndrome de Fraser y dado que no contamos en el país con laboratorio para hacer estudios moleculares para enfermedades genéticas, no logramos confirmarlo por esta metodología. Las intervenciones clínicas y quirúrgicas realizadas permitieron en este caso, ayudar al tratamiento de la paciente la cual egresó del hospital con citas para su seguimiento por diversas especialidades.
Ante recién nacidos que presenten una gama de anomalías congénitas siempre es importante la valoración genética y revisar el caso con el fin de intentar llegar a un diagnóstico o a una sospecha diagnóstica. Debemos trabajar en el desarrollo de un laboratorio nacional de genética que incluya estudios de biología molecular para el diagnóstico de enfermedades y síndromes genéticos.